


Thalassemia symptoms can range from mild to severe and often include fatigue, weakness, pale or yellowish skin, and delayed growth in children. Early recognition of these signs is important for proper diagnosis, timely treatment, and better management of the condition.
What is Thalassemia? A Deep Dive
To fully understand thalassemia symptoms, it is important to first explore the root cause of the condition. Thalassemia is a genetic blood disorder that affects the body’s ability to produce hemoglobin, the protein in red blood cells responsible for carrying oxygen. When hemoglobin production is disrupted, it leads to anemia and a variety of health complications. The severity of thalassemia symptoms depends on the type and genetic variation of the disease.
Genetic Basis and Inheritance Patterns
Thalassemia is an inherited condition, meaning it is passed from parents to children through genes. A child must inherit the defective gene from one or both parents to develop the condition. If only one gene is inherited, the individual is known as a carrier and may not show noticeable thalassemia symptoms, but they can still pass the gene to future generations. When both parents are carriers, the risk of having a child with a more severe form of thalassemia increases significantly.
Different Types of Thalassemia: Alpha and Beta
Hemoglobin is made up of two protein chains: alpha and beta. Thalassemia is classified based on which of these chains is affected. Each type has different levels of severity and produces different ranges of thalassemia symptoms, from mild anemia to life-threatening conditions.
Types of Thalassemia and Severity Overview
| Type | Genetic Defect | Severity Level | Common Impact |
|---|---|---|---|
| Alpha Thalassemia (1 gene missing) | One alpha gene mutation | Very Mild | Usually no symptoms (silent carrier) |
| Alpha Thalassemia (2 genes missing) | Two alpha gene mutations | Mild | Mild anemia |
| Hemoglobin H Disease (3 genes missing) | Three alpha gene mutations | Moderate to Severe | Noticeable anemia and fatigue |
| Hydrops Fetalis (4 genes missing) | All alpha genes affected | Very Severe | Often fatal before or shortly after birth |
| Beta Thalassemia Minor | One beta gene mutation | Mild | Mild anemia, often unnoticed |
| Beta Thalassemia Intermedia | Two genes partially affected | Moderate | Variable symptoms, occasional treatment |
| Beta Thalassemia Major | Both beta genes severely affected | Severe | Requires regular blood transfusions |
Alpha Thalassemia: From Silent Carrier to Severe Conditions
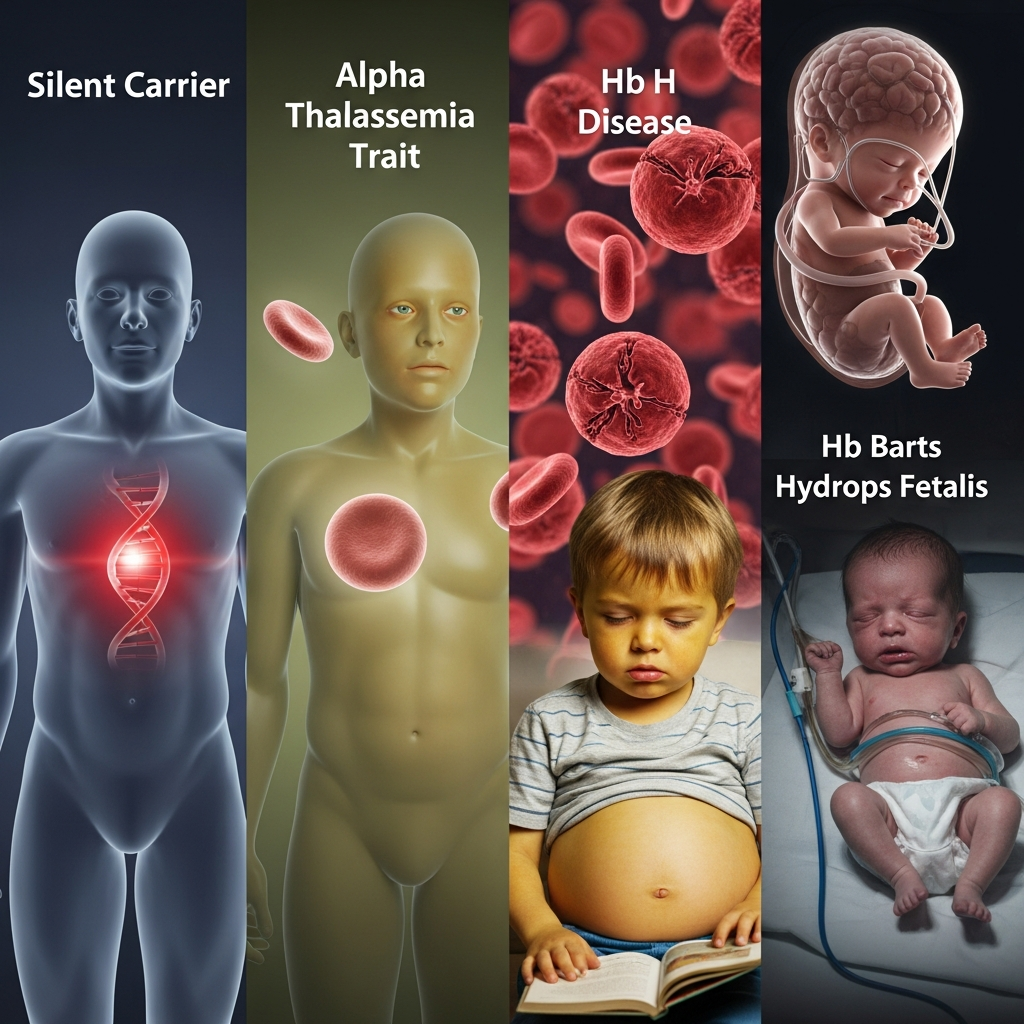 Alpha thalassemia occurs when mutations affect the alpha globin genes. The severity depends on how many genes are missing or defective. Individuals with fewer affected genes may not experience noticeable thalassemia symptoms, while those with more severe genetic defects can develop serious health complications, including moderate to severe anemia and growth issues.
Alpha thalassemia occurs when mutations affect the alpha globin genes. The severity depends on how many genes are missing or defective. Individuals with fewer affected genes may not experience noticeable thalassemia symptoms, while those with more severe genetic defects can develop serious health complications, including moderate to severe anemia and growth issues.
Beta Thalassemia: Minor, Intermedia, and Major
Beta thalassemia affects the beta globin genes and varies widely in severity. People with beta thalassemia minor usually have mild anemia and minimal thalassemia symptoms. However, those with thalassemia major experience severe anemia and require lifelong medical care, including regular blood transfusions and ongoing monitoring to manage complications.
Key Points About Thalassemia Symptoms and Types
- Thalassemia symptoms vary depending on the number of affected genes and the type of thalassemia, ranging from no symptoms to severe, life-threatening conditions.
- The disorder is genetic, meaning early screening and family history play a crucial role in identifying risks and preventing severe cases.
- Alpha and beta thalassemia differ based on which hemoglobin chain is affected, and each type has its own progression and severity level.
- Carriers may not show visible thalassemia symptoms, but they can still pass the condition to their children, making genetic counseling important.
- Severe forms of thalassemia often require long-term treatment such as blood transfusions and specialized medical care to manage symptoms effectively.
Recognizing the Signs: Common Thalassemia Symptoms
Identifying thalassemia symptoms early is essential for timely diagnosis and proper medical care. While mild cases may go unnoticed for years, more severe forms often present symptoms within the first two years of life. These symptoms are primarily linked to anemia and the body’s inability to produce enough healthy red blood cells to carry oxygen efficiently.
Anemia-Related Symptoms: Fatigue, Weakness, and Pallor
The most common thalassemia symptoms are directly related to anemia. Due to a shortage of healthy red blood cells, the body’s tissues do not receive enough oxygen, leading to persistent fatigue and low energy levels. Patients often feel weak even after minimal physical activity and may struggle with daily tasks. A pale or yellowish skin tone (pallor) is another visible sign, along with shortness of breath, dizziness, and occasional headaches. These symptoms can significantly affect quality of life if not properly managed.
Growth and Development Issues in Children
Children with moderate to severe thalassemia symptoms frequently experience delayed growth and slower physical development. The body diverts much of its energy toward producing red blood cells, leaving less energy available for normal growth processes. This can result in shorter height, delayed puberty, and overall developmental delays. Regular health checkups and nutritional support are crucial to monitor progress and ensure early intervention when needed.
Bone Deformities and Brittleness
One of the more serious thalassemia symptoms involves changes in bone structure. As the body tries to compensate for low red blood cell production, bone marrow expands to increase output. This expansion can cause bones—especially in the face and skull—to become wider or misshapen. Over time, bones may become thinner, weaker, and more prone to fractures. These skeletal changes are more common in untreated or severe cases of thalassemia.
Enlarged Spleen and Liver: Causes and Consequences
The spleen plays a vital role in filtering damaged blood cells. In individuals with thalassemia, the spleen works harder than normal to remove defective red blood cells, leading to a condition known as splenomegaly (enlarged spleen). This enlargement can worsen anemia by destroying even healthy blood cells. Similarly, the liver may also become enlarged due to increased workload and iron accumulation. These conditions can cause abdominal discomfort, swelling, and additional complications if not monitored closely.
Jaundice and Dark Urine: What They Mean
Another important sign among thalassemia symptoms is jaundice, which causes a yellowing of the skin and eyes. This occurs due to the rapid breakdown of red blood cells, releasing excess bilirubin into the bloodstream. High bilirubin levels can also affect urine color, making it appear dark or tea-colored. These signs indicate that the body is struggling to process and eliminate damaged blood cells effectively.
Iron Overload: A Silent but Serious Complication
Iron overload is one of the most dangerous long-term thalassemia symptoms and complications. It often results from repeated blood transfusions or increased iron absorption by the body. Excess iron gets stored in vital organs such as the heart, liver, and endocrine glands. Over time, this can lead to serious health issues like heart disease, liver cirrhosis, and hormonal imbalances, including diabetes. Managing iron levels through proper treatment, such as iron chelation therapy, is essential to prevent organ damage.
Summary of Common Thalassemia Symptoms
| Symptom Category | Key Signs |
|---|---|
| Anemia | Fatigue, weakness, pale skin, shortness of breath |
| Growth Issues | Delayed growth, slow development |
| Bone Changes | Facial bone deformities, fragile bones |
| Organ Enlargement | Enlarged spleen and liver |
| Blood Breakdown | Jaundice, dark urine |
| Complications | Iron overload, organ damage |
Diagnosing Thalassemia: The Road to Confirmation
 Accurate diagnosis is essential for developing an effective treatment plan for thalassemia symptoms and preventing long-term complications. Doctors use a combination of clinical evaluation, family history assessment, and advanced laboratory tests to confirm the condition. Early diagnosis is especially important in identifying carriers and managing severe forms of the disease before complications develop.
Accurate diagnosis is essential for developing an effective treatment plan for thalassemia symptoms and preventing long-term complications. Doctors use a combination of clinical evaluation, family history assessment, and advanced laboratory tests to confirm the condition. Early diagnosis is especially important in identifying carriers and managing severe forms of the disease before complications develop.
Initial Blood Tests: CBC and Peripheral Smear
The diagnostic process usually begins with a Complete Blood Count (CBC) test, which measures hemoglobin levels and evaluates the number and type of blood cells. In patients with thalassemia symptoms, CBC often shows low hemoglobin and smaller-than-normal red blood cells (microcytic anemia).
A peripheral blood smear is then performed to closely examine the shape, size, and color of red blood cells under a microscope. Abnormalities such as target cells, irregular shapes, and pale cells strongly suggest the presence of thalassemia. These initial tests help doctors decide whether further specialized testing is required.
Key Diagnostic Tests for Thalassemia
| Test Name | Purpose | What It Shows |
|---|---|---|
| CBC (Complete Blood Count) | Basic blood evaluation | Low hemoglobin, microcytic anemia |
| Peripheral Smear | Cell structure analysis | Abnormal red blood cell shape and size |
| Hemoglobin Electrophoresis | Hemoglobin type analysis | Abnormal hemoglobin patterns |
| HPLC Test | Detailed hemoglobin separation | Accurate identification of hemoglobin variants |
| Genetic Testing | DNA mutation detection | Exact gene mutation causing thalassemia |
| Prenatal Testing (CVS/Amniocentesis) | Fetal diagnosis | Detects thalassemia before birth |
Hemoglobin Electrophoresis and HPLC
To confirm the type of thalassemia, doctors use hemoglobin electrophoresis or High-Performance Liquid Chromatography (HPLC). These tests analyze different types of hemoglobin in the blood and detect imbalances in alpha and beta chains. This step is crucial for understanding the severity of thalassemia symptoms and determining whether the patient has alpha or beta thalassemia. These advanced tests provide a more accurate diagnosis than basic blood tests alone.
Genetic Testing: Confirming the Diagnosis and Carrier Status
Genetic testing is the most definitive method for diagnosing thalassemia. It identifies specific mutations in the genes responsible for hemoglobin production. This test is especially important for detecting carriers who may not show visible thalassemia symptoms but can still pass the gene to their children. Genetic counseling is often recommended alongside testing to help families understand inheritance risks and reproductive options.
Prenatal Diagnosis: Options for Expectant Parents
For couples who are known carriers, prenatal diagnosis offers early detection of thalassemia in the fetus. Procedures such as chorionic villus sampling (CVS) and amniocentesis are used to collect fetal DNA for analysis. These tests are typically performed in early pregnancy and provide critical information about whether the baby has inherited thalassemia. This allows parents to prepare emotionally, medically, and financially for future care decisions.
Managing Thalassemia: Treatment Approaches
While there is currently no simple universal cure for thalassemia, modern medicine offers several effective treatment options to manage thalassemia symptoms, improve quality of life, and prevent complications. Treatment is usually lifelong and tailored based on severity.
Blood Transfusions: A Lifesaving Intervention
Regular blood transfusions are the primary treatment for moderate to severe thalassemia symptoms. These transfusions provide healthy red blood cells, helping improve oxygen delivery throughout the body. As a result, patients experience reduced fatigue, improved growth in children, and better overall physical function. However, transfusions must be carefully managed to avoid complications such as iron overload.
Iron Chelation Therapy: Preventing Iron Overload
Frequent blood transfusions can lead to excess iron accumulation in the body. Iron chelation therapy is used to remove this excess iron and prevent damage to vital organs like the heart and liver. Medications used in this therapy bind to iron and help the body eliminate it through urine or stool. Consistent use is essential, as unmanaged iron overload can worsen thalassemia symptoms and lead to life-threatening complications.
Bone Marrow Transplantation: A Potential Cure
A bone marrow or stem cell transplant is currently the only potential curative treatment for thalassemia. It replaces the patient’s defective bone marrow with healthy stem cells from a compatible donor. This procedure can eliminate thalassemia symptoms completely in some cases, but it is complex, expensive, and requires a perfectly matched donor, often a sibling.
Folic Acid Supplementation
Folic acid is essential for the production of healthy red blood cells. Patients with thalassemia are often prescribed folic acid supplements to support blood formation and reduce the severity of anemia-related thalassemia symptoms. This is a supportive treatment that works alongside other medical therapies.
Lifestyle Modifications and Supportive Care
Managing thalassemia symptoms also involves important lifestyle changes. Patients are often advised to follow a balanced diet rich in essential nutrients like calcium and vitamin D to support bone health. Iron-rich foods and supplements should be avoided unless specifically recommended by a doctor. Maintaining good hygiene is also important, especially for individuals with a weakened immune system or those who have undergone spleen removal. Regular medical checkups, emotional support, and proper infection prevention strategies all contribute to better long-term health outcomes.
Living with Thalassemia: Challenges and Support
 Living with thalassemia is not only about managing physical health but also about coping with emotional, social, and lifestyle challenges. Individuals affected by thalassemia symptoms often require lifelong care, regular monitoring, and strong support systems to maintain a good quality of life. A holistic approach that combines medical treatment, emotional well-being, and social support is essential for long-term stability.
Living with thalassemia is not only about managing physical health but also about coping with emotional, social, and lifestyle challenges. Individuals affected by thalassemia symptoms often require lifelong care, regular monitoring, and strong support systems to maintain a good quality of life. A holistic approach that combines medical treatment, emotional well-being, and social support is essential for long-term stability.
Emotional and Psychological Impact
Living with chronic thalassemia symptoms can have a significant emotional impact on both patients and their families. Frequent hospital visits, regular blood transfusions, and ongoing medical treatments often create stress, anxiety, and emotional fatigue. Many patients also experience feelings of isolation or depression due to long-term illness management.
Children with thalassemia may struggle with self-esteem issues, especially if they experience growth delays or physical complications. Parents may also face emotional pressure while managing the financial and caregiving responsibilities. Seeking psychological counseling, emotional therapy, or peer support can greatly help in coping with these challenges and improving mental well-being.
The Importance of a Multidisciplinary Care Team
Effective management of thalassemia symptoms requires a team-based medical approach. A multidisciplinary care team ensures that all aspects of the disease are properly monitored and treated. This team usually includes hematologists who manage blood-related issues, cardiologists who monitor heart health, endocrinologists who address hormonal imbalances, and nutritionists who guide dietary planning.
Each specialist plays a vital role in preventing complications such as iron overload, organ damage, and growth delays. Regular coordination among these professionals helps create a personalized treatment plan that improves patient outcomes and reduces long-term risks associated with thalassemia.
Support Groups and Resources
Support groups play a crucial role in improving the lives of people living with thalassemia symptoms. These groups provide a safe space where patients and families can share experiences, challenges, and coping strategies. Emotional encouragement from others facing similar situations can significantly reduce stress and feelings of isolation.
In addition, many national and international organizations offer educational materials, financial assistance programs, and awareness campaigns to support the thalassemia community. Access to these resources helps patients stay informed and better manage their condition while also advocating for improved healthcare services.
The Future of Thalassemia Treatment: Research and Innovations
Ongoing medical research is bringing new hope for better management and potential cures for thalassemia symptoms. Scientists and healthcare professionals are continuously working on advanced therapies that target the root cause of the disease rather than just managing its effects.
Gene Therapy: A Promising Frontier
Gene therapy is one of the most exciting developments in thalassemia research. This approach involves correcting the defective gene responsible for abnormal hemoglobin production by inserting a healthy gene into the patient’s stem cells. Early clinical trials have shown promising outcomes, with some patients experiencing significant improvement and reduced dependency on blood transfusions. This innovation could potentially transform the future treatment of thalassemia symptoms.
New Drug Developments
Researchers are also developing new medications aimed at improving red blood cell production and reducing complications associated with thalassemia symptoms. Some drugs focus on stimulating healthy hemoglobin production, while others help regulate iron levels more effectively. These emerging treatments are still under clinical testing but show strong potential for improving long-term disease management and reducing treatment burden.
Early Detection and Prevention Programs
Preventive healthcare plays a major role in reducing the impact of thalassemia symptoms worldwide. Early detection through newborn screening programs and carrier testing helps identify the condition at an early stage. Public health awareness campaigns are also increasing education about genetic risks, especially in high-risk regions.
These programs empower individuals and couples to make informed decisions about family planning and genetic counseling. By focusing on prevention and early diagnosis, healthcare systems can significantly reduce the number of severe thalassemia cases in future generations.
 Receiving a diagnosis of thalassemia can be emotionally overwhelming for both patients and their families. However, gaining a clear understanding of the condition is one of the most powerful steps toward effective management. Early recognition of thalassemia symptoms allows for timely medical intervention, which can significantly reduce complications and improve long-term health outcomes. With proper awareness, education, and support, individuals can take control of their condition rather than feeling controlled by it.
Receiving a diagnosis of thalassemia can be emotionally overwhelming for both patients and their families. However, gaining a clear understanding of the condition is one of the most powerful steps toward effective management. Early recognition of thalassemia symptoms allows for timely medical intervention, which can significantly reduce complications and improve long-term health outcomes. With proper awareness, education, and support, individuals can take control of their condition rather than feeling controlled by it.