


Stem cell transplant for thalassemia is a potentially curative treatment that replaces a patient’s faulty bone marrow with healthy stem cells from a compatible donor. It helps the body produce normal hemoglobin and reduces or eliminates the need for regular blood transfusions. This procedure is most effective in younger patients with a matched donor and is considered one of the most advanced treatment options for severe thalassemia.
Understanding Thalassemia: Types, Symptoms, and Current Treatments
To appreciate how a stem cell transplant for thalassemia works, you first need to understand the mechanics of this condition. Thalassemia is a genetic blood disorder that occurs when the genes responsible for producing the alpha or beta chains of hemoglobin are mutated or missing. This leads to reduced or abnormal hemoglobin production, causing chronic anemia and long-term health complications.
Different Types of Thalassemia
Thalassemia is typically categorized by severity, and understanding these types is essential for proper thalassemia diagnosis and treatment planning.
Thalassemia Minor
Individuals usually inherit one defective gene. Most people experience only mild anemia and may not require regular treatment. In many cases, it is discovered incidentally during a routine hemoglobin panel test.
Thalassemia Intermedia
This moderate form causes more noticeable symptoms than the minor type. Patients may experience fatigue and anemia that can worsen during illness, pregnancy, or physical stress. Some may require occasional blood transfusions depending on severity.
Thalassemia Major
Also known as Cooley’s anemia, this is the most severe form of the disease. Patients require regular blood transfusions, iron chelation therapy, and continuous medical monitoring. In such cases, advanced treatment options like stem cell transplant for thalassemia may be considered as a potential curative approach.
Recognizing the Symptoms of Thalassemia
 Early diagnosis plays a critical role in managing this condition effectively. Symptoms often appear within the first two years of life in severe cases. Children may show extreme fatigue, pale or yellowish skin, poor appetite, and slow physical growth.
Early diagnosis plays a critical role in managing this condition effectively. Symptoms often appear within the first two years of life in severe cases. Children may show extreme fatigue, pale or yellowish skin, poor appetite, and slow physical growth.
As the body tries to compensate for low hemoglobin production, the bone marrow becomes overactive, which can lead to bone deformities, especially in the face and skull. This is why early screening and regular hemoglobin panel testing are essential for identifying the severity of the disease and planning appropriate thalassemia treatment.
In more advanced cases, complications such as organ enlargement, delayed puberty, and iron overload may develop, requiring long-term medical management and sometimes consideration of curative options like stem cell transplant for thalassemia. If you want to learn more about identifying these signs early, read our comprehensive Thalassemia Symptoms Guide.
Traditional Thalassemia Management
Historically, treating severe thalassemia has relied heavily on regular blood transfusions. These transfusions provide the body with the healthy red blood cells it cannot make itself. For detailed information on treatment schedules, explore our guide on Thalassemia Blood Transfusion Frequency.
While transfusions save lives, they introduce a massive influx of iron into the bloodstream. The body cannot naturally eliminate this excess iron, leading to dangerous buildup in the heart and liver. Therefore, patients must also undergo iron chelation therapy, a treatment designed to bind and remove excess iron from the body.
The Science Behind Stem Cell Transplantation
A stem cell transplant for thalassemia offers a completely different treatment approach compared to traditional therapies. Instead of repeatedly managing symptoms with blood transfusions and medications, this procedure aims to correct the root cause by rebuilding the body’s ability to produce healthy blood cells. This makes it one of the most advanced options in modern thalassemia treatment.
What are Stem Cells?
Stem cells are unique, unspecialized cells that have the ability to develop into different types of cells in the body. In the case of thalassemia, doctors primarily focus on hematopoietic stem cells (HSCs). These cells are found in the bone marrow and are responsible for producing red blood cells, white blood cells, and platelets.
In patients with severe thalassemia, these stem cells are defective, which leads to abnormal hemoglobin production. A stem cell transplant for thalassemia works by replacing these faulty cells with healthy ones from a compatible donor, offering the possibility of long-term disease correction.
How a Stem Cell Transplant Works
The goal of the procedure is to replace the patient’s defective hematopoietic stem cells with healthy donor cells that can produce normal blood components. This process involves several carefully controlled stages.
Donor Matching
The first and most critical step is Human Leukocyte Antigen (HLA) typing. This genetic testing helps identify a donor whose tissue markers closely match the patient’s. A strong HLA match reduces the risk of rejection and increases the success rate of the stem cell transplant for thalassemia.
Conditioning Regimen
Before the transplant, the patient undergoes a conditioning phase using high-dose chemotherapy or sometimes radiation therapy. This step is essential to destroy the existing faulty bone marrow and suppress the immune system, preventing it from attacking the newly transplanted cells. It is a crucial part of preparing the body for successful engraftment.
Infusion of Stem Cells
The actual transplant procedure is similar to a blood transfusion. Healthy donor stem cells are infused into the patient’s bloodstream through an intravenous line. This step is generally painless and is considered the “transplant day.”
Engraftment Process
After infusion, the stem cells travel through the bloodstream and settle in the bone marrow. Over the next few weeks, they begin to grow and produce healthy red blood cells, white blood cells, and platelets. This important stage is called engraftment, and it marks the beginning of a new, healthy blood-forming system.
A successful engraftment is the key indicator that the stem cell transplant for thalassemia is working effectively and may eventually eliminate the need for lifelong blood transfusions and iron chelation therapy.
Types of Stem Cell Transplants for Thalassemia
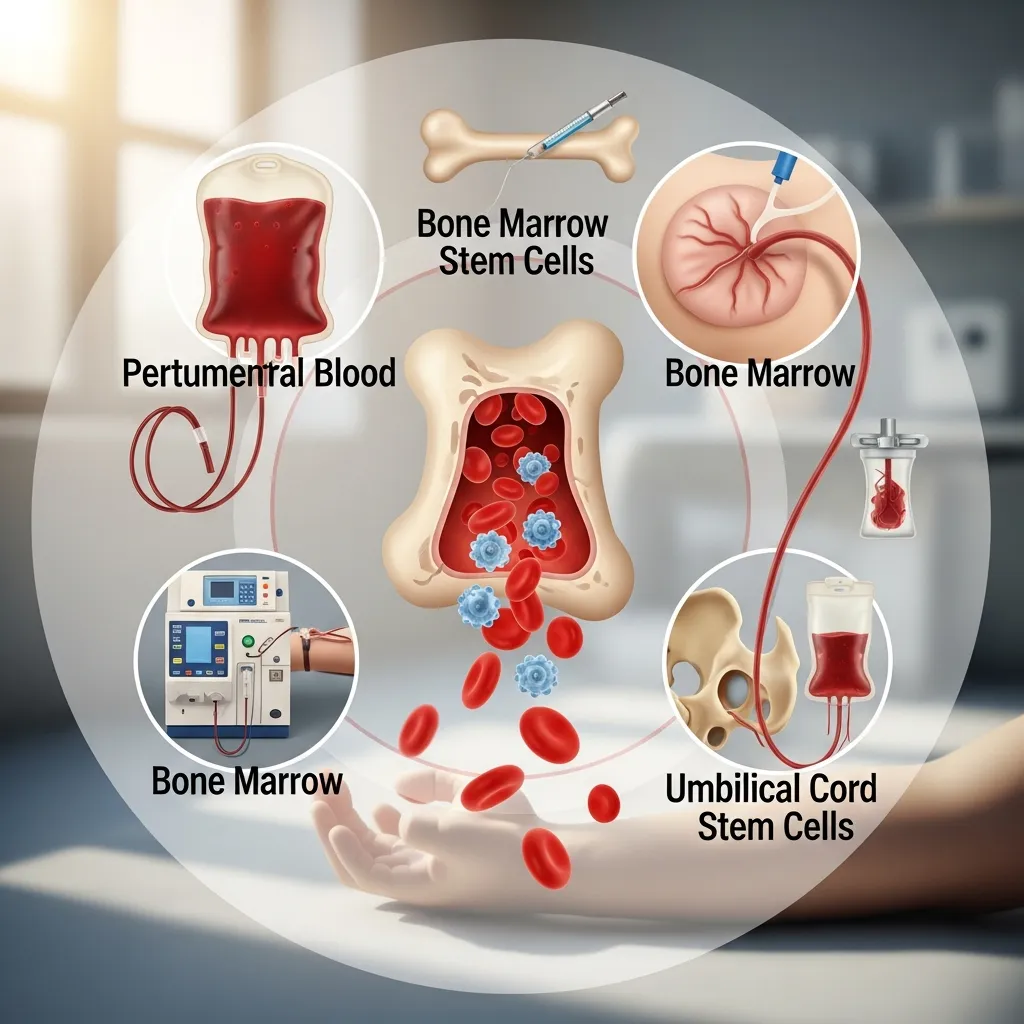 Doctors select the specific type of transplant based on donor availability, patient health, and the severity of the disease.
Doctors select the specific type of transplant based on donor availability, patient health, and the severity of the disease.
Allogeneic Stem Cell Transplant
This is the most common approach for thalassemia. It involves using stem cells from a healthy donor.
- Matched Sibling Donors: A brother or sister with the exact same HLA type is the ideal donor. This scenario offers the highest success rates and lowest complication risks.
- Matched Unrelated Donors (MUD): If a sibling match isn’t available, doctors search global registries for an unrelated individual who shares the same HLA markers.
- Haploidentical Transplants: Sometimes, a half-matched donor (usually a parent) is used. Advanced medical techniques have made these transplants much safer and more successful in recent years.
Cord Blood Transplants
Umbilical cord blood is rich in hematopoietic stem cells. Families can choose to bank cord blood when a healthy child is born. If an older sibling has thalassemia, these stored cells can be used for a transplant. Cord blood stem cells are highly adaptable, meaning they do not require as perfect an HLA match as adult bone marrow cells.
Autologous Transplants
Traditionally, autologous transplants—using the patient’s own stem cells—were not viable for genetic disorders like thalassemia. However, emerging gene therapies are changing this. Scientists are currently testing ways to extract a patient’s own stem cells, correct the defective thalassemia gene in a laboratory, and infuse them back into the body.
The Stem Cell Transplant Process: A Step-by-Step Guide
Undergoing a stem cell transplant is a major medical journey that requires careful planning and specialized care.
Pre-Transplant Evaluation and Preparation
Long before the actual procedure, the patient undergoes rigorous testing. Doctors evaluate heart, liver, and lung function to ensure the body can withstand the intense conditioning treatments. During this phase, patients also address any existing iron overload through aggressive chelation therapy.
The Conditioning Phase
Patients are admitted to a specialized hospital unit. For several days, they receive carefully calculated doses of chemotherapy. This phase can be physically taxing, causing side effects like nausea, hair loss, and extreme fatigue.
The Transplant Procedure
The transplant itself is remarkably straightforward. The patient receives the donor stem cells through a central venous catheter. The infusion usually takes a few hours, and patients are closely monitored for any immediate allergic reactions.
Post-Transplant Recovery and Monitoring
The period immediately following the transplant is highly critical. Because the conditioning regimen wipes out the immune system, the patient is exceptionally vulnerable to infections. They must remain in a sterile hospital environment for several weeks while waiting for engraftment to occur. Doctors monitor blood counts daily, providing blood transfusions and antibiotics as needed until the new marrow starts functioning.
Benefits and Risks of Stem Cell Transplant for Thalassemia
Like any major medical intervention, a stem cell transplant carries both profound rewards and significant risks.
Potential Benefits: Long-Term Cure
The primary benefit is the potential for a complete cure. A successful transplant eliminates the need for lifelong blood transfusions and iron chelation therapy. Patients experience improved energy levels, normal growth and development, and a vastly improved quality of life. They can live free from the heavy medical burden that severe thalassemia imposes.
Potential Risks and Complications
The procedure does involve serious risks that require careful consideration.
- Graft-versus-Host Disease (GvHD): This occurs when the newly transplanted immune cells recognize the patient’s body as foreign and attack it. GvHD can range from a mild skin rash to a severe, life-threatening condition affecting the liver and digestive tract.
- Infections: The prolonged period of immune suppression makes patients highly susceptible to bacterial, viral, and fungal infections.
- Organ Toxicity: The high doses of chemotherapy used during conditioning can cause temporary or permanent damage to organs like the liver, lungs, or heart.
- Relapse: In some cases, the new stem cells fail to take hold, or the patient’s original, defective bone marrow grows back, causing the thalassemia to return.
Eligibility Criteria and Patient Selection
A stem cell transplant is not suitable for everyone with thalassemia. Medical teams carefully evaluate several factors before recommending this path.
Factors Influencing Candidacy for Transplant
- Age and Overall Health: Outcomes are generally best for younger patients, particularly children who have not yet accumulated significant organ damage from iron overload. Older patients or those with severe liver or heart complications face higher risks during the conditioning phase.
- Thalassemia Type and Severity: Transplants are typically reserved for patients with thalassemia major who depend on regular blood transfusions to survive.
- Availability of a Suitable Donor: Finding an exact HLA-matched donor significantly influences the decision to proceed. Without a suitable match, the risks of complications like GvHD increase substantially.
Life After a Stem Cell Transplant for Thalassemia
Recovery does not end when the patient leaves the hospital. It takes time for the immune system to fully rebuild and for the body to heal.
Long-Term Follow-Up Care
Patients require close monitoring for months and even years following a transplant. They undergo frequent blood tests to ensure the new bone marrow is producing healthy cells. Doctors also monitor for late-onset complications, including chronic GvHD and organ function issues.
Managing Post-Transplant Complications
If complications like GvHD arise, patients may need long-term immunosuppressive medications. Additionally, those who accumulated significant iron before the transplant may still require phlebotomy (blood removal) or continued chelation therapy until their iron stores normalize.
Quality of Life and Outlook
For the vast majority of successful transplant recipients, the long-term outlook is excellent. Once fully recovered, they can attend school, pursue careers, and engage in physical activities without the constraints of severe anemia. The psychological relief of overcoming a chronic, life-threatening illness is also immense.
Advancements and Future Directions in Thalassemia Treatment
 The medical landscape is constantly evolving, offering new hope for patients who may not currently qualify for a traditional transplant.
The medical landscape is constantly evolving, offering new hope for patients who may not currently qualify for a traditional transplant.
Gene Therapy and Gene Editing
Gene therapy holds incredible promise for the future. Instead of relying on donor cells, this approach modifies the patient’s own stem cells to produce functional hemoglobin. You can read more about the latest research on Gene Therapy for Thalassemia from the National Institutes of Health. These therapies bypass the need for an HLA-matched donor and eliminate the risk of GvHD.
Ongoing Clinical Trials
Researchers are continuously running clinical trials to make transplants safer and more effective. Current studies focus on developing less toxic conditioning regimens, improving engraftment rates for haploidentical transplants, and finding better ways to prevent and treat GvHD.
Improving Donor Search and Matching Technologies
Global networks are making it easier for patients to find unrelated donors. Organizations like the World Marrow Donor Association work tirelessly to expand international donor registries, increasing the odds that a patient with thalassemia will find their perfect match, regardless of their ethnic background.